引言
一般来讲,我们想要使用TCGA数据,大概有三种方法,一是直接从GDC官网或官方下载工具gdc-client下载文件后自行处理,二是使用数据库如UCSC Xena或Firehouse,三是使用TCGAbiolinks R包自动下载并处理。
从官网下载并不麻烦,但是第一是需要选取非常多的自定义选项,第二是网络环境不好会容易中断,对于初学者倒是一个非常好的了解生物信息学的途径,但遇到批量化处理需求的时候就会难以进行。其后是数据库法,数据库虽然方便,但是并不会随着官网的更新变动,如 GDC Xena Hub 最后一次更新时间是 2019-08-28 , Firehose 更是停留在了更遥远的 2016_01_28 ……
那么, 如果我需要批量下载的话, 难道我需要一个个的从网页加入Cart获取mata吗, 我不要……
幸好,已经有人造了非常好用的轮子,当然可以轻松学习一下用起来啦。
TCGAbiolinks 包是从TCGA数据库官网接口下载数据的R包。它的一些函数能够轻松地帮我们下载数据和整理数据格式。其实就是broad研究所的firehose命令行工具的R包装!
需要注意的是,2022年TCGA数据库进行了一次比较大的更新,其中包括了数据格式的变动,因此 TCGAbiolinks 也必须随之更新到最新版。下面,正式开始。
效果展示
可获得文件如下:
- TCGA转录组数据原始文件(tsv)及临床原始文件(xml), 均附带清单
- 表达矩阵表格(可选
"counts", "fpkm", "tpm")
- 分组文件
- 临床数据, 其中包含生存数据
过程
下载
首先是更新最新版的 TCGAbiolinks 包, 我使用的办法是使用Clash获得本地代理后对 R session 进行代理流量转发, 而后直接运行 BiocManager::install("TCGAbiolinks") , 选择所有包更新, 很顺利的就更新好了.
1
2
3
4
| Sys.setenv(http_proxy="http://127.0.0.1:7890")
Sys.setenv(https_proxy="http://127.0.0.1:7890")
Sys.setenv(all_proxy="socks5://127.0.0.1:7890")
BiocManager::install("TCGAbiolinks")
|
其中, 127.0.0.1 是本机(local_host)的ip地址, 而7890是Clash的默认端口.
安装成功后,就可以开始使用了。
如, 运行 TCGAbiolinks:::getGDCprojects()$project_id 获取各个癌种的项目id, 总计有74个.
1
2
3
4
5
6
7
| library(TCGAbiolinks)
TCGAbiolinks:::getGDCprojects()$project_id
TCGAbiolinks:::getGDCprojects()$project_id %>% length()
|
如需获取TCGA癌症数据, 可以使用正则表达式获取开头带有 TCGA 的项目.
1
2
3
4
5
6
7
8
9
| projects <- TCGAbiolinks::getGDCprojects()$project_id
projects <- projects[grepl("^TCGA", projects, perl = TRUE)]
projects
projects %>% length
|
TCGA的33种癌症简写对应全称可在UCSC Xena查到.
查看一个项目内的可下载文件:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
| TCGAbiolinks:::getProjectSummary("TCGA-COAD")
|
下载并提取mRNA的表达矩阵, 其中数据类别 data.category 是 Transcriptome Profiling 代表转录组数据; 数据类型 data.type 是 Gene Expression Quantification , 代表表达谱数据; 定量方式 workflow.type 是 STAR - Counts , 内含了 Counts / FPKM 和 FPKM-UQ 三种数据类型.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
|
query <- GDCquery(
project = project,
data.category = "Transcriptome Profiling",
data.type = "Gene Expression Quantification",
workflow.type = "STAR - Counts"
)
head(query$results[[1]], n=10) %>% View()
GDCdownload(query, method = "api", files.per.chunk = 100)
|
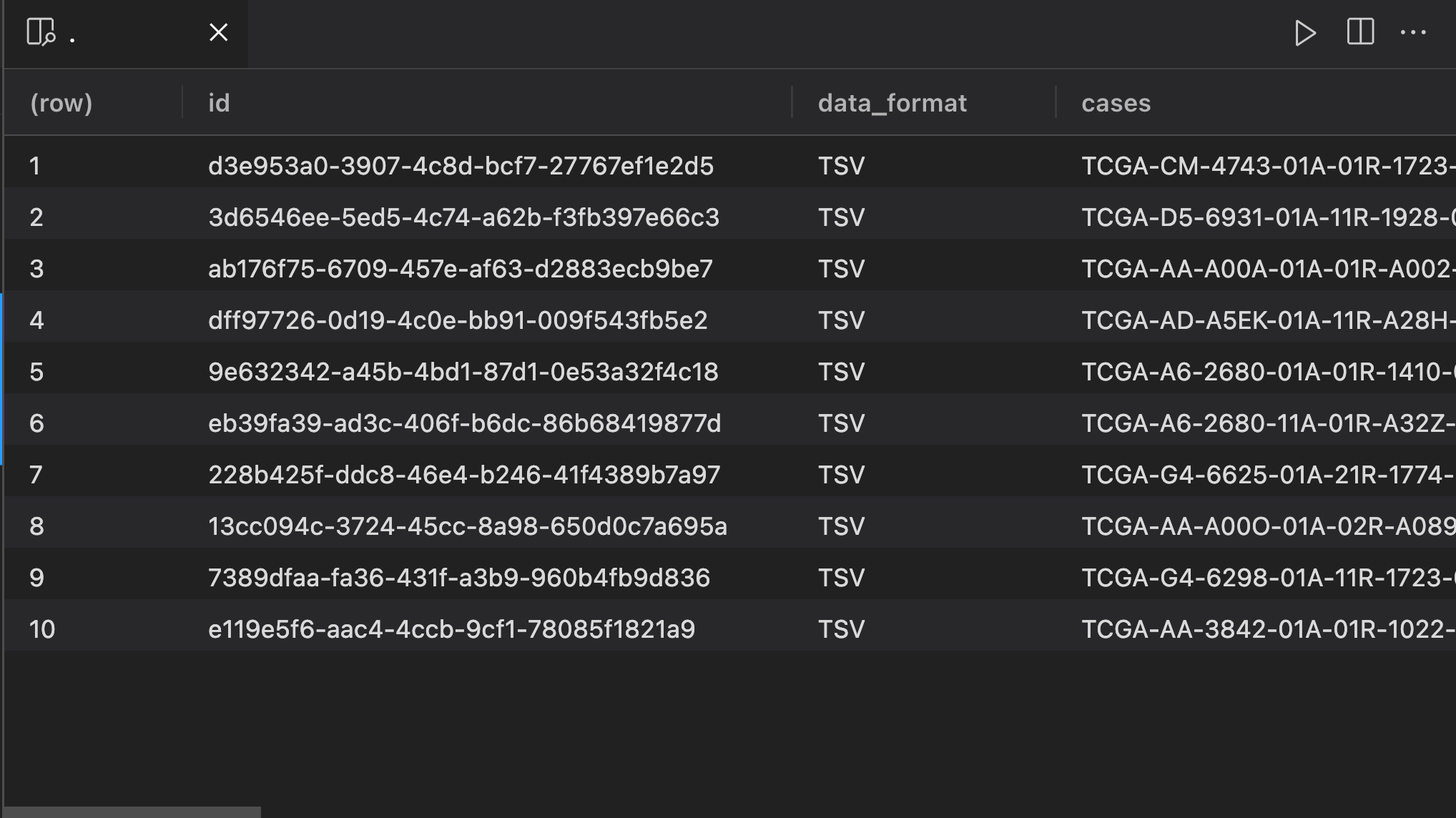
这里出现了一个意外… 我的硬盘提示空间不足了.. 在这个内存动不动64的年代, 我这个硬盘总共200g的可怜人实惨.. 可见 GDCprepare 函数需要强大的内存和硬盘空间, 我的本地电脑是做不到的, 因此继续使用老方案进行数据处理. 目前为止, 通过 TCGAbiolinks 进行数据下载的目的已经圆满达到.
首先保存 MANIFEST 文件以供后续分析:
1
2
| shelfEnvironment(paste(imput_dir, "GDCdata", project, sep = "/"), path = root_dir)
write.csv(query$results[[1]], file = paste0(project, "_MANIFEST.csv"))
|
shelfEnvironment 函数来源于 obgetDEGs 包, 可使用 devtools::install_github('sandy9707/obgetDEGs') 命令安装, 函数的作用是将目标文件夹设定为工作目录, 如果该目录不存在便创建. 详情可参考GitHub - sandy9707/obgetDEGs.
该函数的应用场景是:当需要在R中读取或写入数据时,需要指定存储数据的文件夹路径。但在执行R代码时,可能需要将当前工作目录更改为存储数据的文件夹路径。如果文件夹不存在,需要创建文件夹。这时, shelfEnvironment 函数可以帮助我们检查并创建文件夹,使得数据可以正常读取或写入。
表达谱数据处理
清空环境, 读取MANIFEST信息, 特别是需要样本名和文件夹名.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
|
rm(list = ls())
(root_dir <- sub("/code.+", "", rstudioapi::getSourceEditorContext()$path))
source(paste(root_dir, "code", "prepare.R", sep = "/"))
project <- "TCGA-COAD"
shelfEnvironment(paste(imput_dir, "GDCdata", project, sep = "/"), path = root_dir)
json <- read.csv(paste0(project, "_MANIFEST.csv"))
case_names <- json[, "cases"]
filedir_in_json <- json[, "file_id"]
|
选择提取部分
1
2
3
4
5
6
7
|
extract_type <- c("counts", "fpkm", "tpm")[1]
extract_num <- switch(extract_type,
"counts" = 4,
"fpkm" = 8,
"tpm" = 7
)
|
开始提取, 原理是进入每一个文件夹并提取某列, 再结合基因类型, 并去重.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
|
matrix_MMRF_list <- list()
shelfEnvironment(paste(imput_dir, "GDCdata", project,
"/harmonized/Transcriptome_Profiling/Gene_Expression_Quantification",
sep = "/"
), path = root_dir)
for (i in 1:length(filedir_in_json)) {
setwd(paste0("./", filedir_in_json[i]))
matrix_MMRF_list[[i]] <- read.table(
file = list.files(pattern = ".tsv"),
header = F, fill = TRUE
)[, extract_num]
setwd("../")
}
setwd(paste0("./", filedir_in_json[i]))
probematrix <- read.table(list.files(pattern = ".tsv"),
header = F, fill = TRUE
)[, 2:3]
setwd("../")
matrix_MMRF <- do.call(cbind, matrix_MMRF_list)
matrix_MMRF <- cbind(probematrix, matrix_MMRF)
tibble_MMRF <- tibble::as_tibble(matrix_MMRF)
colnames(tibble_MMRF) <- c("gene_name", "gene_type", str_sub(case_names, 1, 16))
duplicated(colnames(tibble_MMRF), fromLast = TRUE) %>% table()
tibble_MMRF <- tibble_MMRF[, !duplicated(colnames(tibble_MMRF), fromLast = TRUE)]
duplicated(colnames(tibble_MMRF), fromLast = TRUE) %>% table()
|
提取蛋白编码基因并将基因名保留转换行名.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
|
pcg <- c(
"protein_coding", "IG_V_gene", "IG_D_gene", "IG_J_gene",
"IG_C_gene", "TR_V_gene", "TR_D_gene", "TR_J_gene", "TR_C_gene"
)
mrna_exprset <- tibble_MMRF %>%
dplyr::filter(gene_type %in% pcg) %>%
dplyr::select(-gene_type) %>%
dplyr::distinct(gene_name, .keep_all = TRUE) %>%
tibble::column_to_rownames("gene_name")
|
通过TCGA样本命名规则筛选需求样本并将对照组前置.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
|
mrna_exprset %>%
select(-matches("[^(01)]A$|[^(11)]A$")) %>%
ncol()
mrna_exprset %>%
select(matches("[01]1[A]$")) %>%
ncol()
mrna_exprset <- mrna_exprset %>%
select(matches("[01]1A$")) %>%
select(-matches(".-0[1-9][A]$"), everything())
ncol(mrna_exprset)
|
写出表达矩阵, 特征列表和分组列表.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
|
shelfEnvironment(paste(imput_dir, "GDCdata", project, sep = "/"),
path = root_dir
)
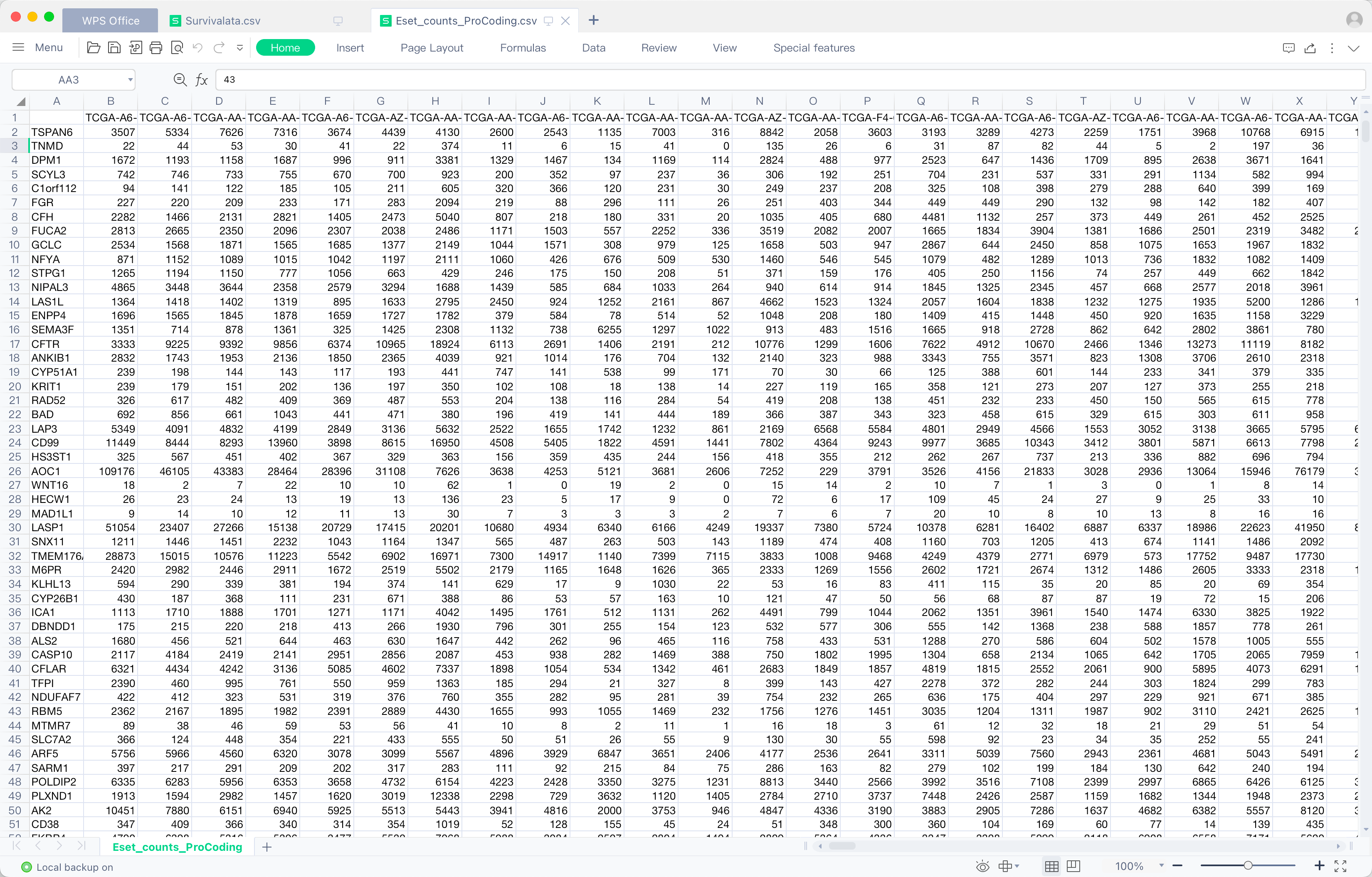
write.csv(mrna_exprset, paste0("Eset_", extract_type, "_ProCoding.csv"), row.names = T)
write.table(rownames(mrna_exprset), "feature_list.csv",
row.names = FALSE, quote = FALSE, col.names = FALSE
)
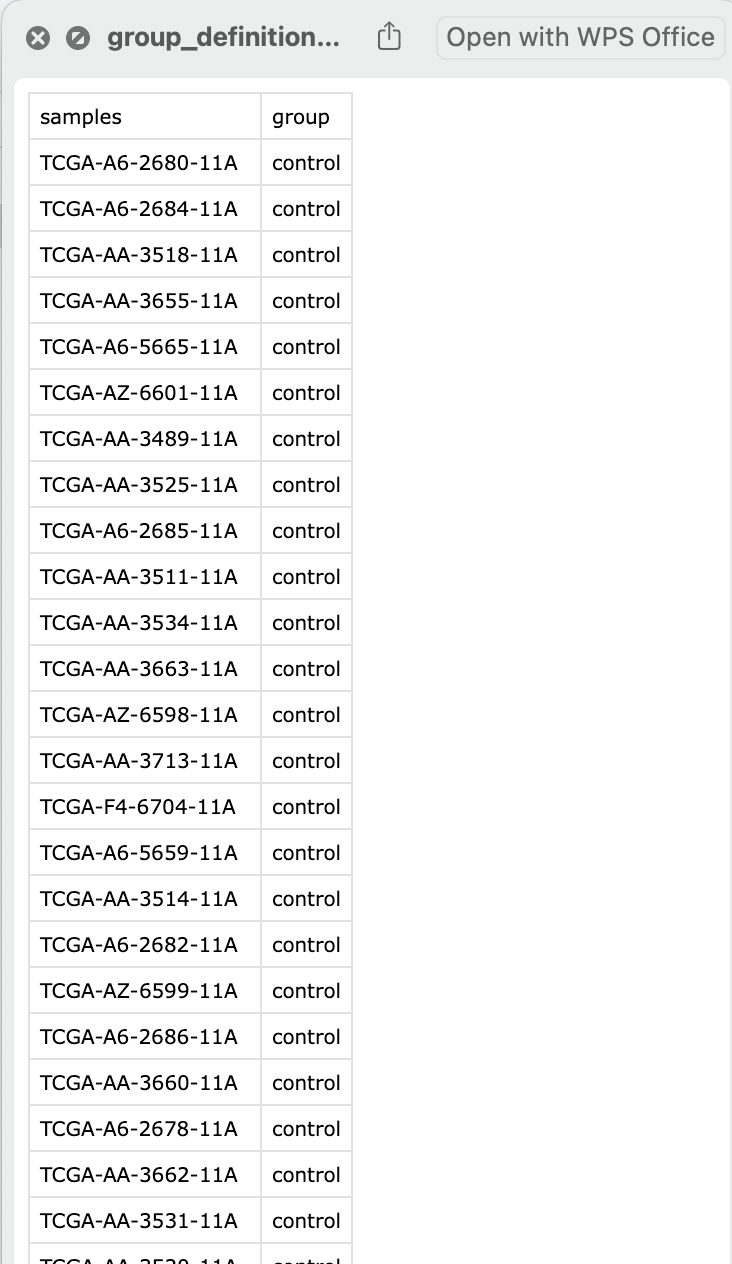
samples <- colnames(mrna_exprset)
group_definition_matrix <- data.frame(
samples,
group = ifelse(grepl("11A$", samples), "control", "test")
)
write.csv(group_definition_matrix,
file = "group_definition_matrix.csv", row.names = F
)
|
1
2
3
4
5
6
7
8
9
| sapply(projects, function(project){
query <- GDCquery(project = project,
data.category = "Clinical",
file.type = "xml")
GDCdownload(query)
clinical <- GDCprepare_clinic(query, clinical.info = "patient")
saveRDS(clinical,file = paste0(project,"_clinical.rds"))
})
|
临床数据下载及处理
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
|
project
sapply(projects, function(project) {
query <- GDCquery(
project = project,
data.category = "Clinical",
file.type = "xml"
)
GDCdownload(query)
clinical <- GDCprepare_clinic(query, clinical.info = "patient")
})
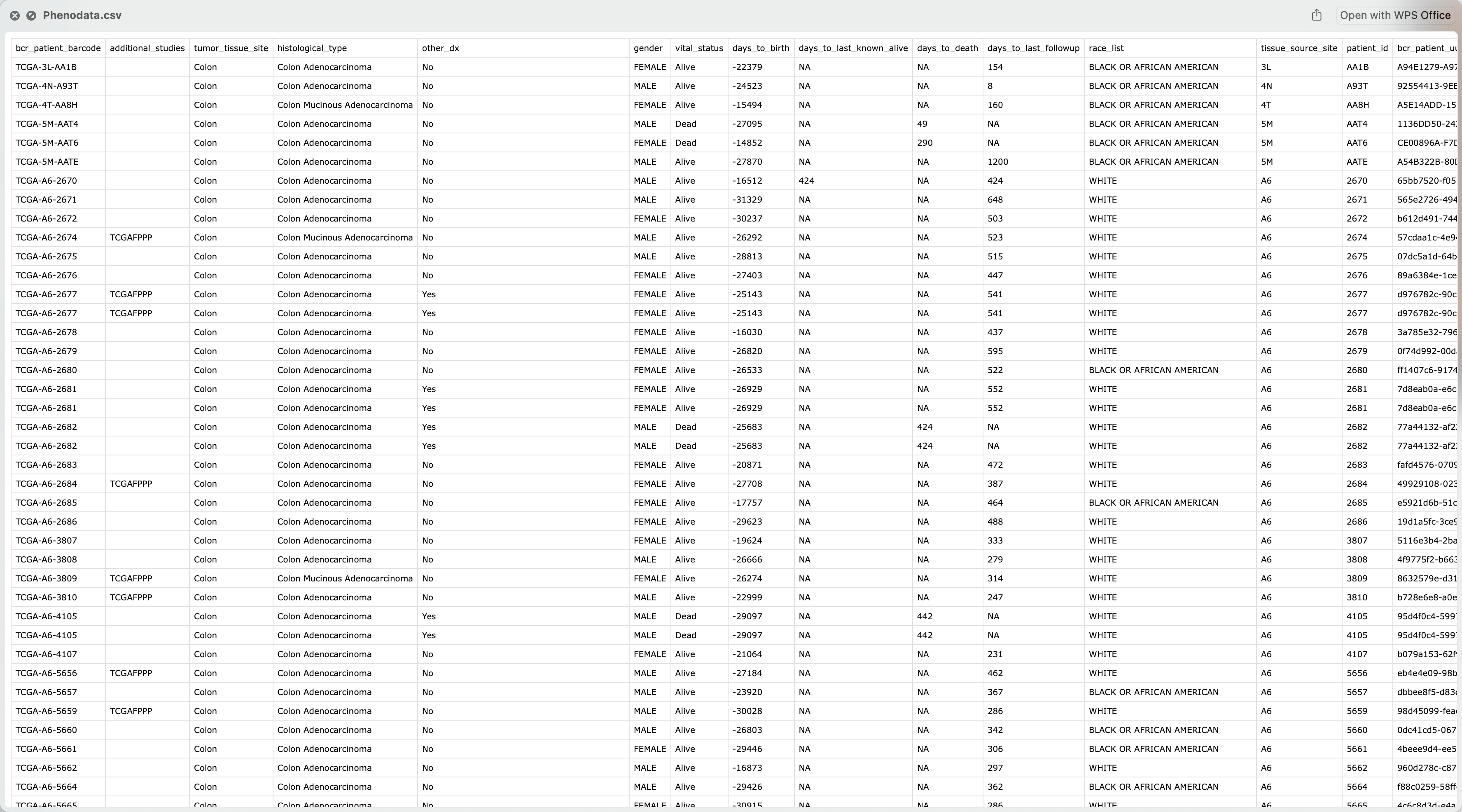
write.csv(clinical, paste0("Phenodata.csv"), row.names = F)
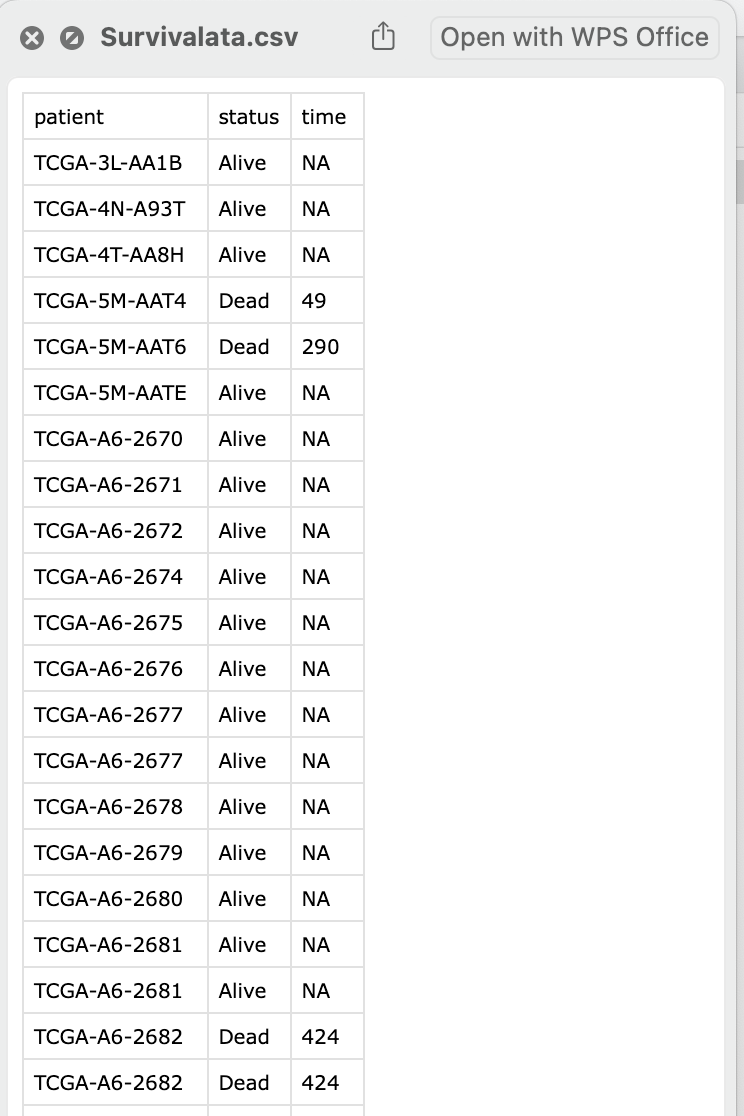
col_survivaldata <- setNames(
data.frame(clinical[, c("bcr_patient_barcode", "vital_status", "days_to_death")]),
c("patient", "status", "time")
)
write.csv(col_survivaldata, paste("Survivalata.csv", sep = "/"), row.names = F)
|
总结
如想获得本文的全套代码以及工作环境, 只需要在公众号私信发送 TCGA数据下载整理 就可以获得.
引用
- GDC官网
- 官方下载工具gdc-client
- UCSC Xena
- Firehouse
- TCGAbiolinks
- TCGA数据下载—TCGAbiolinks包参数详解 - 腾讯云开发者社区-腾讯云
- 新版TCGAbiolinks包学习:批量下载数据新版TCGA数据 - mdnice 墨滴
- TCGA / 癌症简称 / 缩写 / TCGA癌症中英文对照
- GitHub - sandy9707/obgetDEGs
- TCGA样本命名规则

