引言
之前介绍过 如何使用TCGAbiolinks下载TCGA数据并整理 , 那么如果手动整理又该如何呢?
下面以 miRNA 数据整理为例示范.
效果展示
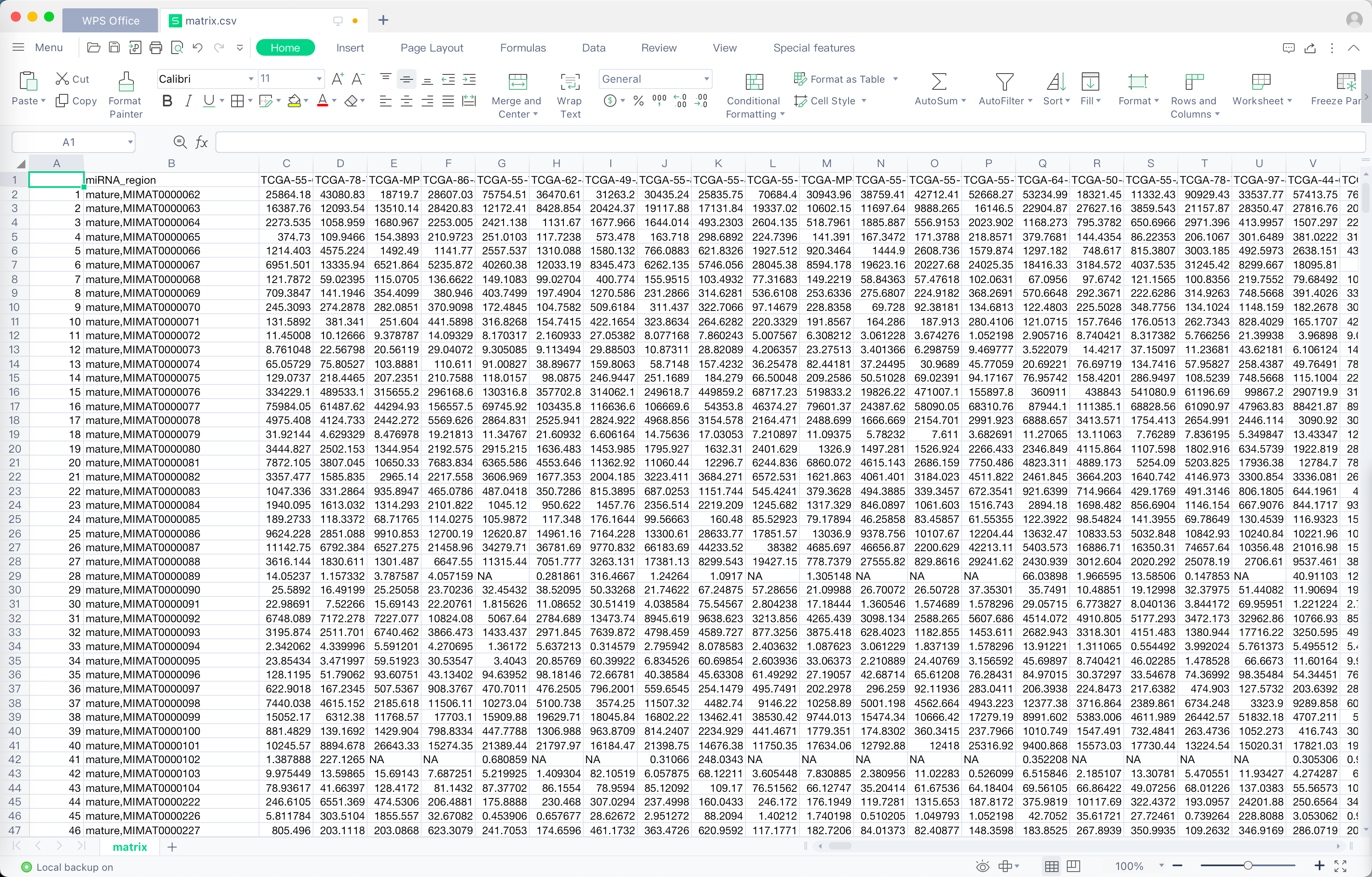
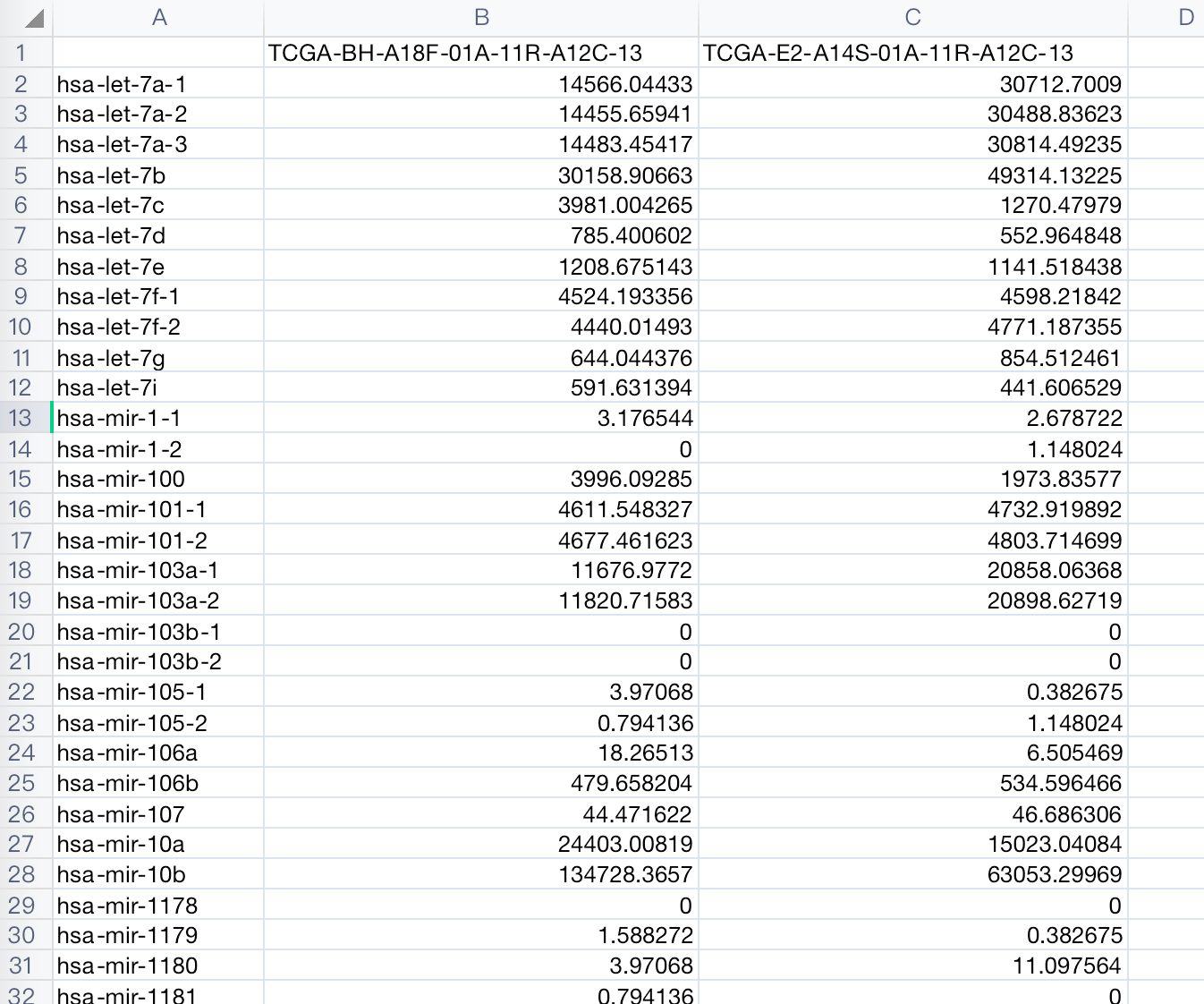
过程
输入文件
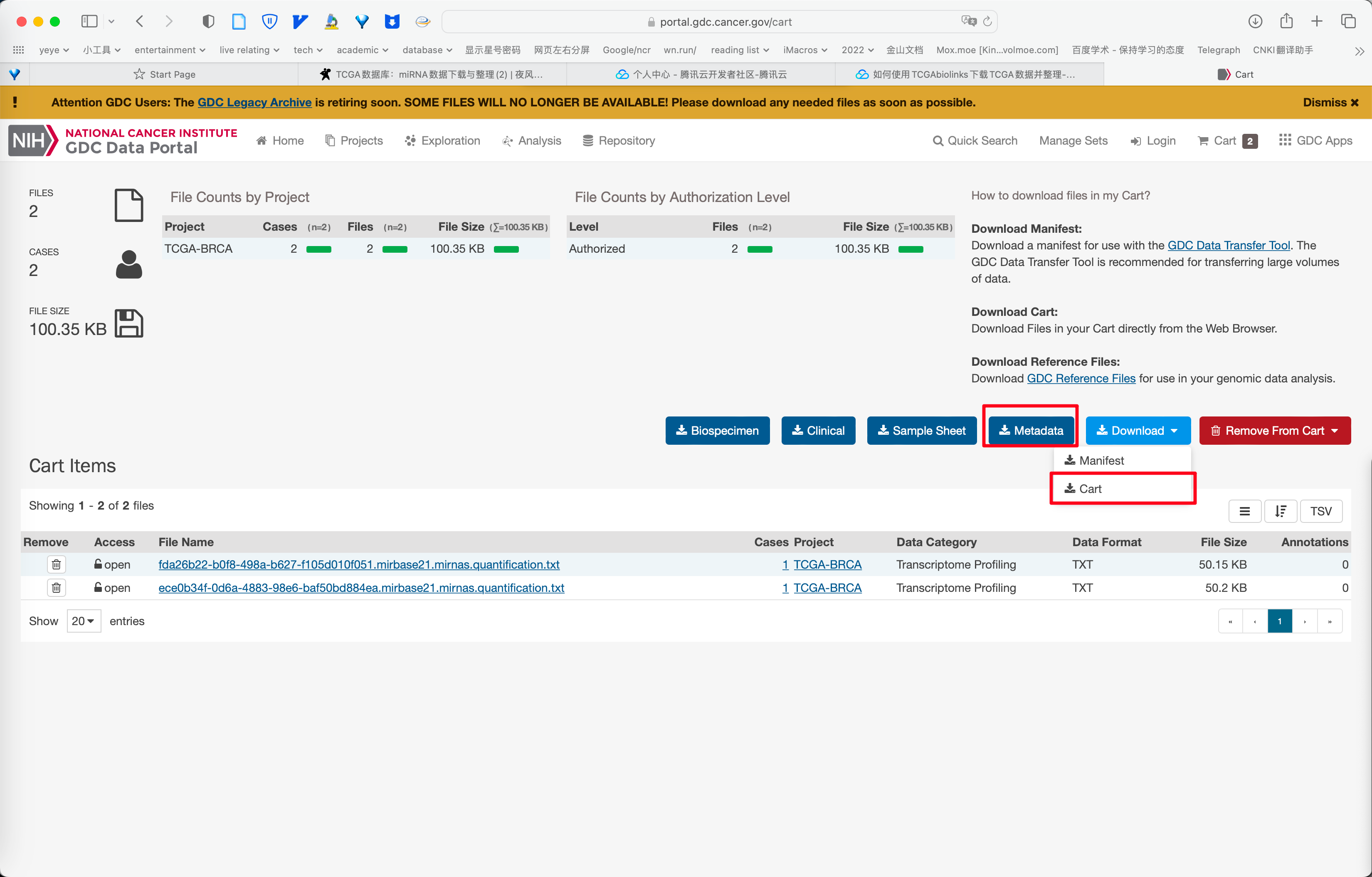
随便下载一些数据, 下载格式选择 Metadata 和 Cart .
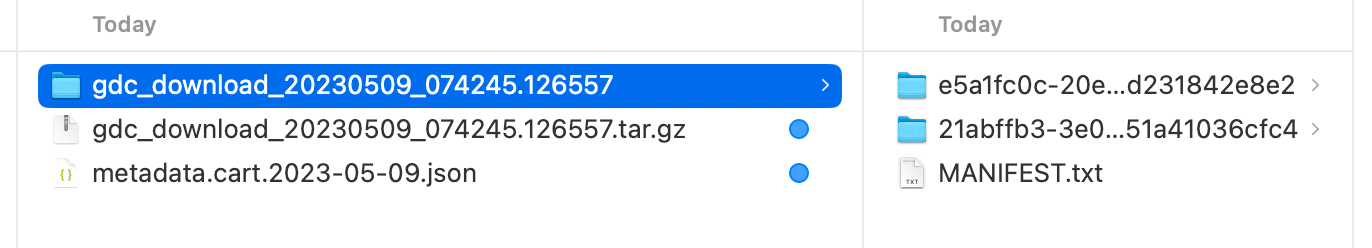
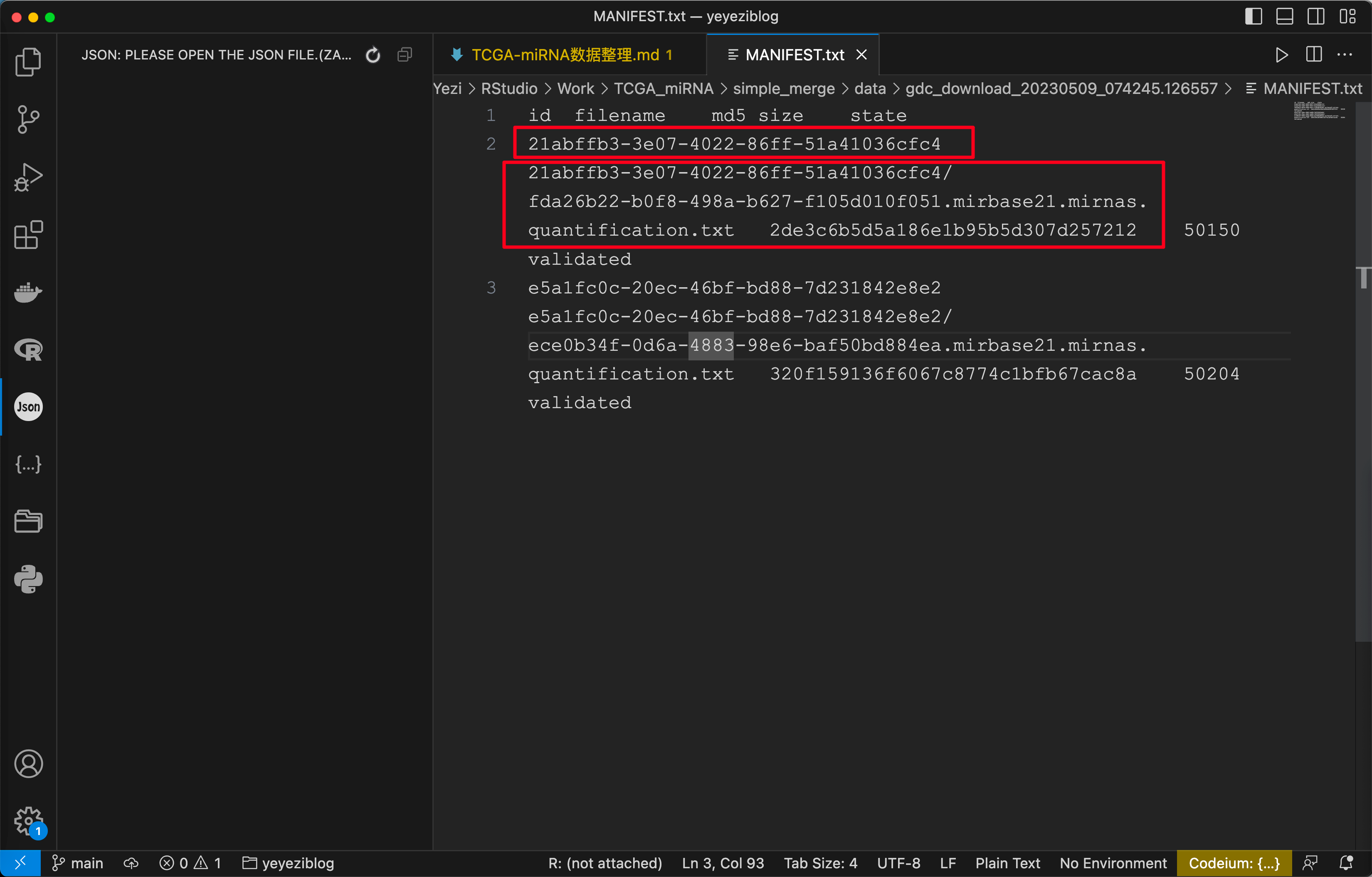
下载得到一个 Metadata 的 json 文件和一个包含全部数据的压缩包, 解压可得到 MANIFEST.txt 和一堆文件夹.
观察可得 Metadata.json 包含了所需读入文件名和样本的 TCGA Submitter Id .
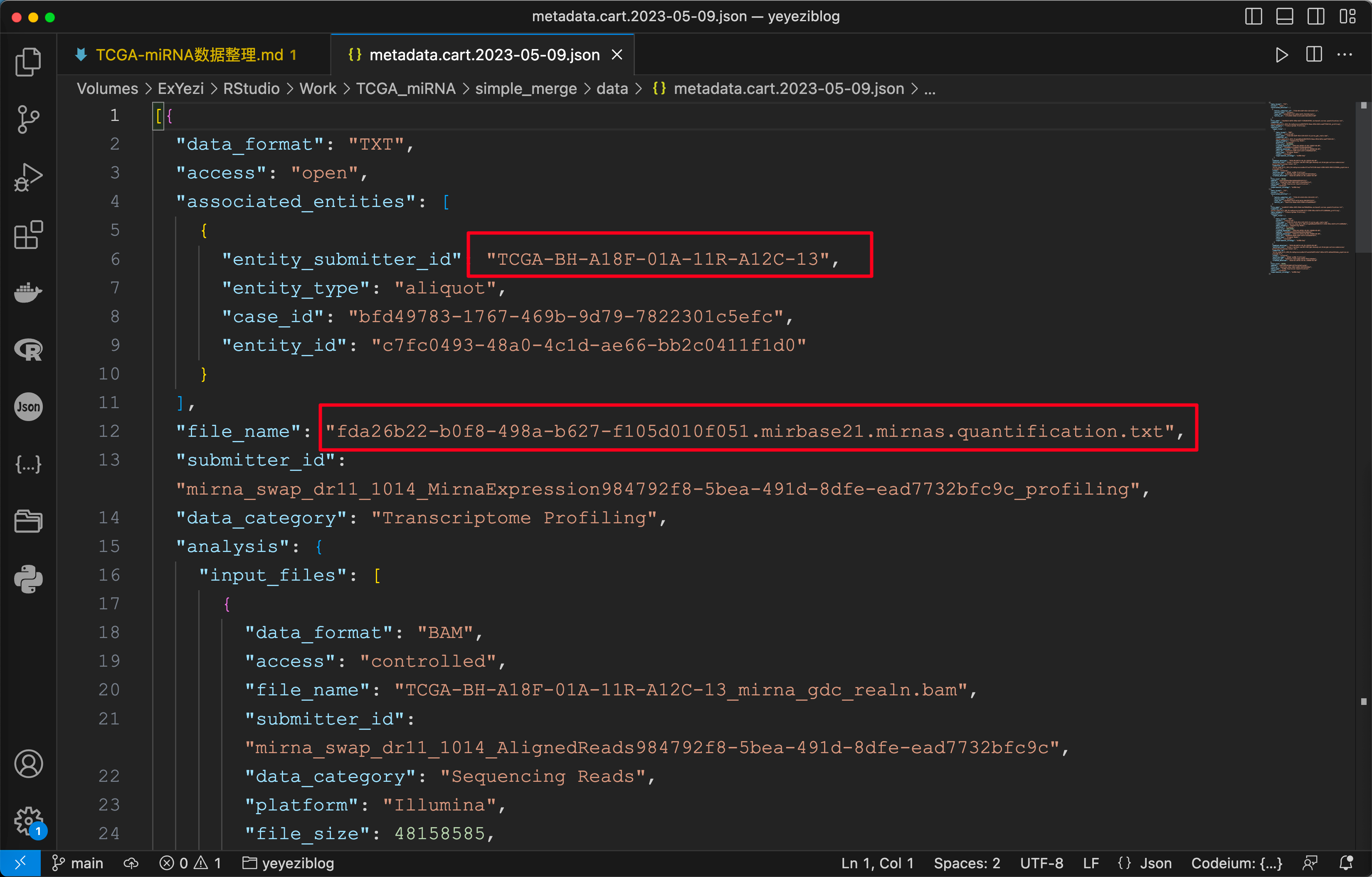
同样对 MANIFEST.txt 观察可得其中包含了所需读入文件名和文件所在的文件夹.
因此就可以使用 R 对已下载数据做简单处理.
R代码整理
配置工作环境
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
|
rm(list = ls())
(root_dir <- sub("/main.*", "", rstudioapi::getSourceEditorContext()$path))
shelf_folder <- function(folder_name, parent_folder = ".") {
target_folder <- file.path(parent_folder, folder_name)
if (!file.exists(target_folder)) {
dir.create(target_folder, recursive = TRUE)
}
setwd(target_folder)
return(target_folder)
}
data_folder <- shelf_folder("data", root_dir)
results_folder <- shelf_folder("results", root_dir)
library(librarian)
shelf(dplyr, stringr, quiet = TRUE)
shelf_folder("data", root_dir)
|
此代码可以在脚本所在目录创建 data 和 results 文件夹,并提供了一个快速创建并设定工作目录的 function。
将所有的TCGA下载文件及解压后的文件夹放入 data 中。
处理json文件
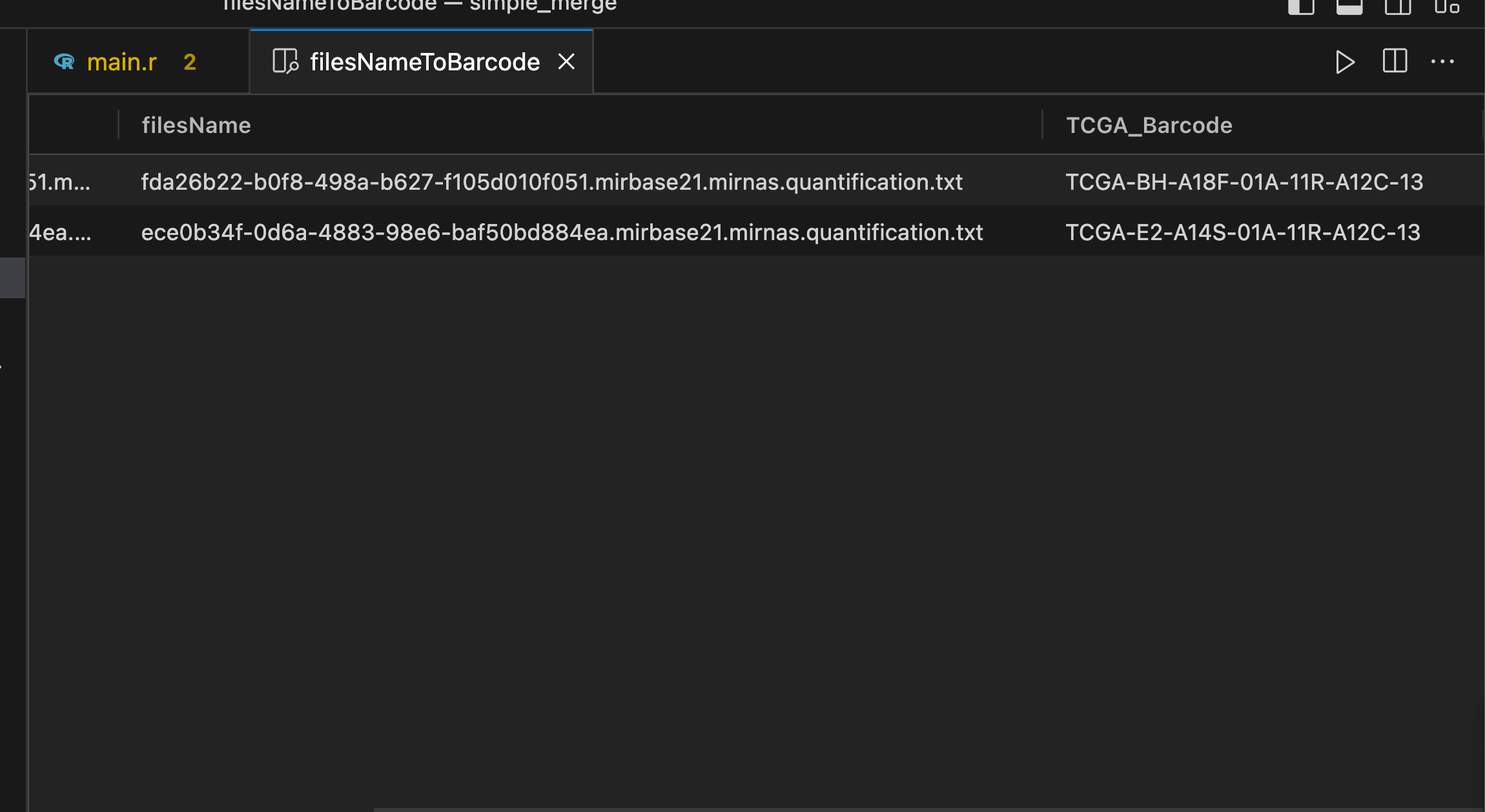
之后使用代码对json文件做处理得到所需读入文件名和样本 TCGA Submitter Id 之间的对应关系, 代码来源于 TCGA数据库:miRNA数据下载与整理(2) | 夜风博客 .
1
2
3
4
5
6
7
8
9
10
11
12
13
|
shelf(jsonlite, tidyr)
list.files(data_folder)
json_file <- read_json(paste0(data_folder, "/metadata.cart.2023-05-09.json"))
length(json_file)
filesNameToBarcode <- data.frame(filesName = c(), TCGA_Barcode = c())
for (i in 1:length(json_file)) {
TCGA_Barcode <- json_file[[i]][["associated_entities"]][[1]][["entity_submitter_id"]]
file_name <- json_file[[i]][["file_name"]]
filesNameToBarcode <- rbind(filesNameToBarcode, data.frame(filesName = file_name, TCGA_Barcode = TCGA_Barcode))
}
(rownames(filesNameToBarcode) <- filesNameToBarcode[, 1])
|
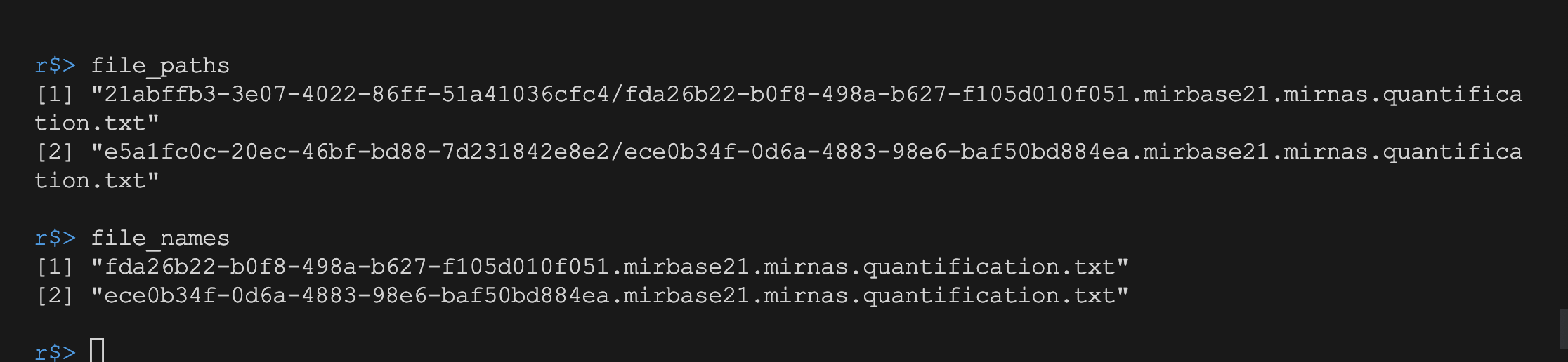
处理MANIFEST文件
处理MANIFEST文件得到所需读入文件名和文件所在文件夹的对应关系.
1
2
3
4
5
|
gdc_folder <- shelf_folder("gdc_download_20230509_074245.126557", data_folder)
MANIFEST <- read.table(paste0(gdc_folder, "/manifest.txt"), sep = "\t", header = TRUE)
file_paths <- MANIFEST[, "filename"]
file_names <- sub(".*/", "", file_paths)
|
依次读入文件并合并
依次读入文件并合并,原理是创建一个空列表,再利用for循环依次从文件中提取值并填充。之后使用do。call命令对列表内全部项进行cbind处理。需要注意的是,cbind函数要求合并矩阵行名保持一致。
其中,合并数据为counts或RPM由read.table后的提取列1或2决定。
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
|
i <- 1
matrix_list <- list()
for (i in 1:length(file_paths)) {
file_path <- file_paths[i]
matrix_list[[i]] <- read.table(paste0(gdc_folder, "/", file_path),
sep = "\t", header = TRUE, row.names = 1
)[, 2, drop = FALSE]
}
matrix <- do.call(cbind, matrix_list)
colnames(matrix) <- filesNameToBarcode$TCGA_Barcode[match(file_names, filesNameToBarcode$filesName)]
head(matrix)
write.csv(matrix, file = paste0(results_folder, "/matrix.csv"))
|
根据反馈修改
小伙伴反馈表示 miRNA 数据并不一定存在一致的行名, 因此更换思路为按行名分组求和后合并矩阵, 缺失值以 Na 填充.
核心代码为(读入过程和合并过程):
读入过程使用了group_by函数进行分组,使用了summarise_all(sum)进行组内相加。
1
2
3
| summarized_data <- data %>%
group_by(miRNA_region) %>%
summarise_all(sum)
|
合并过程使用了for循环对第二列之后的列依次以left_join函数组合到第一列上.
1
2
3
4
| matrix <- matrix_list[[1]]
for (i in 2:length(matrix_list)) {
matrix <- left_join(matrix, matrix_list[[i]], by = "miRNA_region")
}
|
以下为完整代码(可改”reads_per_million_miRNA_mapped”为”read_count”):
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
|
i <- 1
matrix_list <- list()
for (i in 1:length(file_paths)) {
file_path <- file_paths[i]
data <- read.table(paste0(gdc_folder, "/", file_path),
sep = "\t", header = TRUE
)[, c("reads_per_million_miRNA_mapped", "miRNA_region"), drop = FALSE]
summarized_data <- data %>%
group_by(miRNA_region) %>%
summarise_all(sum)
matrix_list[[i]] <- summarized_data
}
matrix <- matrix_list[[1]]
for (i in 2:length(matrix_list)) {
matrix <- left_join(matrix, matrix_list[[i]], by = "miRNA_region")
}
nrow(matrix)
head(matrix)
length(colnames(matrix))
colnames(matrix)[2:length(colnames(matrix))] <- filesNameToBarcode$TCGA_Barcode[match(file_names, filesNameToBarcode$filesName)]
head(matrix)
write.csv(matrix, file = paste0(results_folder, "/matrix.csv"))
|
结论
miRNA的前体可能对应多个成熟的miRNA,比如hsa-let-7a-1,有两个对应的成熟体,MIMAT0000062(hsa-let-7a-5p)和MIMAT0004481(hsa-let-7a-3p)。这里的值是对所有成熟体miRNA求和的结果。
如 TCGA数据库:miRNA数据下载与整理(2) | 夜风博客 文中所说, miRNA的前体可能对应多个成熟的miRNA, 因此还需要使用miRBaseVersions.db包对miRNA_region进行转换, 过程在原文非常清晰, 在此不在赘述.
事实上这种提取方法不局限于miRNA数据, 同样可对普通的转录组数据使用, 感兴趣的朋友可以自行摸索.
本文的完整代码可在公众号回复关键词获得(请复制粘贴):
TCGA-miRNA数据整理
引用
- TCGA数据库:miRNA数据下载与整理(2) | 夜风博客
- Codeium

